Friday, June 19, 2026, was celebrated worldwide as World Sickle Cell Awareness Day.
This global initiative highlights the urgent need to eliminate healthcare disparities and improve access to life-saving treatments across all income levels and regions.
Fittingly then, the theme for this year’s observance is “Closing the Survival Gap: Equity in Sickle Cell Care."
In Ghana, we celebrate two of our most selfless medical luminaries, Professors Kwaku Ohene-Frempong and Felix Konotey-Ahulu, who devoted their entire lives to improving the lives and well-being of people living with sickle cell disease globally.
What is sickle cell, and how is it inherited?
Blood in each of us is made up of different cells, one of which is the red blood cells. The function of the red blood cells is to transport oxygen from the lungs to various parts of the body, where it is utilised for normal systemic function.
The main protein that transports oxygen in the red blood cells is called haemoglobin. This specialised protein is a paired one with each pair inherited from each parent.
The normal haemoglobin is called haemoglobin A. There are other types of haemoglobin which are abnormal, such as haemoglobins S, C, F, etc.
These abnormal haemoglobin types result from subtle differences in the building blocks of proteins, called the amino acid sequence of the protein structure.
Sickle cell disease is an inherited disorder in which one inherits two abnormal haemoglobins, one from each parent, one of which must be the haemoglobin S type. Sickle cell anaemia results when both abnormal haemoglobin chains are of the S type.
The different types of sickle cell disease are determined by the specific combination of abnormal haemoglobin genes a person inherits.
These include haemoglobin SC (HbSC), sickle cell–fetal haemoglobin (HbSF), sickle beta-plus thalassaemia (HbS/β+) and sickle beta-zero thalassaemia (HbS/β0).
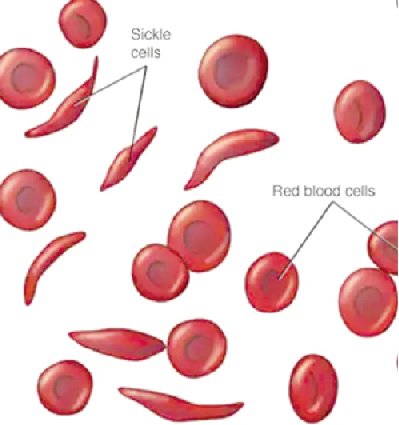
The underlying abnormality is that in sickle cell disease, the red blood cells (RBC's) are abnormally shaped.
The cells change from the normal round, doughnut shape to an elongated shape of a sickle, or letter "C." Sickled cells become stiff and pointed, unlike normal RBC's, which move easily through small blood vessels.
The sickle shape predisposes these red cells to get stuck in narrow blood vessels and block blood flow. This can cause severe pain and lead to organ damage because the cells are deprived of oxygen.
Sickled red blood cells have a shorter-than-normal lifespan, which leads to a low red blood cell count called anaemia. Normal red blood cells can live up to approximately 120 days, whereas abnormal sickled cells live for only 10 to 20 days.
Children affected with the disease have inherited a sickle cell gene from each parent. Someone who inherits only one sickle cell gene and a normal gene from the other parent will have the sickle cell trait.
People with the sickle cell trait do not have sickle cell disease or exhibit any signs of the disorder, but they can still pass the disease on to their children.
When two people who have the trait marry, there is a 25 per cent chance that their offspring will have sickle cell disease.
When one parent carries the trait, and the other actually has the disease, the odds increase to 50 per cent for a child born to them to inherit the disease.
It is worth noting that a one in four (25 per cent) chance of having a child with the disease when both parents have the trait simply means that with each pregnancy, there is a 25 per cent chance of having a child with sickle cell disease.
It is not that when you have four children, one of them will have the disease.
It could be that if a couple who both have the traits decide to have three children, all three could have the disease, all three could escape the disease, or they may have a mix of the various combinations.
Symptoms of sickle cell disease
Symptoms of sickle cell disease can vary and range from mild to severe. The symptoms also vary depending on whether the abnormal haemoglobin combination is SS, SC or other types.
Those with the SS combination tend to have more severe manifestations of the disease compared to the SC or SF counterparts.
Most children with sickle cell disease have anaemia and may develop one or more of the following conditions and symptoms as part of the disorder:
Hand-foot syndrome (dactylitis) - Painful swelling of the hands and feet in children below 2 years of age.
Infection - People with sickle cell anaemia are at an increased risk of certain bacterial infections.
Painful crises - Pain can occur in any part of the body and may be caused by cold, dehydration or stress.
Splenic sequestration crises - The spleen becomes enlarged by trapping the abnormal RBC's. This leads to fewer cells in the general circulation and worsens the anaemia, with sometimes fatal consequences.
Acute chest syndrome - trapping of sickled red blood cells in the lungs.
Aplastic crisis - Bone marrow temporarily slows its production of RBC's due to infection.
Stroke - Poor blood flow can occur in the brain when sickle cells block small blood vessels. This may lead to a stroke.
Other possible complications of sickle cell disease include leg ulcers, bone and joint damage, gallstones, kidney damage, painful, prolonged erections in males (priapism), eye damage and delayed growth.
Treatment of sickle cell disease
Besides a bone marrow transplant, there is no known cure for sickle cell disease. Transplants can become a complicated procedure.
However, even after a bone marrow transplant, the disease may be cured in the individual, but it can still be transmitted to his/her children as the defect is locked in the genes.
Even without a cure, people with sickle cell disease do lead very normal lives. Certain types of medication can be taken to help manage the pain and other complications.
Immunisations help prevent infections. Infants and young children usually require two daily doses of penicillin until they are at least five years old.
They should also be fully immunised with all regular childhood vaccinations.
Daily vitamin supplements, such as folic acid, which help in new red cell production, are routinely given.
Other drugs increase fetal haemoglobin (HbF), a relatively better haemoglobin. An example is Hydroxyurea, which interferes with the sickling process, making red blood cells less sticky.
This helps to decrease the number and intensity of painful episodes and other complications.
Gradually, more people living with the condition are gaining access to hydroxyurea in Ghana, including children. People with SCD should also drink plenty of fluids to avoid dehydration and get a lot of rest.
Early detection of this condition is priceless and available in some selected tertiary hospitals.
The introduction of our expanded programme of immunisation of the pneumococcal and meningococcal vaccines has been a big plus for this condition.
Detection of risk factors for stroke is done with transcranial Doppler ultrasound.
Newborn screening is currently ongoing at certain hospitals, including Korle Bu Teaching Hospital, aimed at early detection.
Perhaps the single most important measure is educating the populace about knowing their sickle cell status while in school, before they start developing feelings for the opposite sex.
In this respect, certain organisations have been helpful, such as the Sickle Cell Condition Advocates (SICCA).
However, we all have a part to play. So, wherever we are, let's play our part in creating awareness of sickle cell disease on our social media handles.
The writer is a member of the Paediatric Society of Ghana and the Director of Medical Affairs at Korle Bu Teaching Hospital.
